Following prolonged therapy with afatinib, many independent afatinib-resistant
http://caspasesignaling.com/foll ... fatinib-resistant-c
Right after prolonged treatment method with afatinib, many independent afatinib-resistant clones emerged that persisted and proliferated even within the presence of two ?M afatinib. From these, we established 14 various independent clones that acquired resistance to afatinib. A few of the drug responses with the representative resistant clones are CYP17 Inhibitors shown in Fig. 1B. To keep track of no matter if the acquisition of resistance was irreversible, we cultured all of these clones in standard development medium with no the RTK inhibitor. These clones remained resistant to afatinib soon after incubation in standard development medium for quite a few months , indicating that these cells underwent an irreversible alter, potentially involving chemically stable physical alterations in their genetic space. The signature exon 19 deletion of parental PC9 cells remained unaltered in all of the r . Full surgical resection of tumors or liver transplantation is only probable in a minority of patients; for patients with innovative condition, the prognosis is extremely poor, with an overall median survival of only some months. Response rates to classical chemotherapy are reduced, and also with combination regimens, long lasting remission has remained elusive . Thus, there is a solid need for added therapeutic possibilities.
Lately, rationally created, molecularly targeted medicines have grown to be out there. These agents are built to target growth or survival pathways hyper-activated in cancer cells. Tyrosine kinases are considered to become exceptional molecular oncology targets Dabigatran due to the fact they transduce growth and survival signals and therefore are hyper-activated in many, if not all, human malignancies . The ErbB receptor tyrosine kinase family members, comprising EGFR and ErbB2, -3, and -4, has become of central interest during the development of targeted anticancer strategies. Trastuzumab , a monoclonal antibody against ErbB2, is efficiently being utilized in individuals with ErbB2-overexpressing breast cancer, and overexpression of ErbB2 through gene amplification is really a excellent predictor of favorable response . Several preclinical and clinical research have addressed the efficacy of EGFR-targeting agents, including tyrosine kinase inhibitors , for example gefitinib and erlotinib , likewise as monoclonal anti-EGFR antibodies, like cetuximab, for that treatment of non-small cell lung cancer , head and neck cancer, colon carcinoma, glioblastoma, together with other tumors . Even though NSCLC sufferers with activating mutations inside the kinase domain of EGFR respond favorably to EGFR TKIs, top to their approval for this subset of malignancies, the molecular basis figuring out the response of tumor cells to EGFR-targeting drugs in other settings is only partially understood and it is talked about controversially.
|
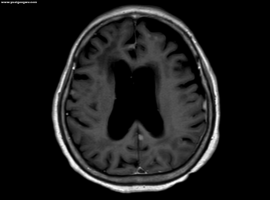 L861Q多发脑转和脑膜转使用佐利替尼
患者是我妈妈,今年71岁,治疗经过如下:
2023-6-7 确诊右肺上叶肺腺癌伴多发淋巴结、
L861Q多发脑转和脑膜转使用佐利替尼
患者是我妈妈,今年71岁,治疗经过如下:
2023-6-7 确诊右肺上叶肺腺癌伴多发淋巴结、
 疾病控制率93.8%,首次亮相的这款肺
作者:seacat
2025年世界肺癌大会(WCLC 2025)报道了KRAS突变的最新进展,包括二代KR
疾病控制率93.8%,首次亮相的这款肺
作者:seacat
2025年世界肺癌大会(WCLC 2025)报道了KRAS突变的最新进展,包括二代KR
 三线击败化疗后,扭过头又获批二线,
作者:Tony
EGFR突变非小细胞肺癌(NSCLC)更易出现在亚洲人群中,其发生率高达40%-50
三线击败化疗后,扭过头又获批二线,
作者:Tony
EGFR突变非小细胞肺癌(NSCLC)更易出现在亚洲人群中,其发生率高达40%-50
 肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双
肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双
 多彩人生 | 抗癌重生之路,她亲手写
讲述者:刘乃荣整理者:pear
48岁本命年那年,我平静的生活迎来转折点——确诊乳腺癌
多彩人生 | 抗癌重生之路,她亲手写
讲述者:刘乃荣整理者:pear
48岁本命年那年,我平静的生活迎来转折点——确诊乳腺癌





 显身卡
显身卡



